درمان تالاسمی با فناوری ویرایش ژن
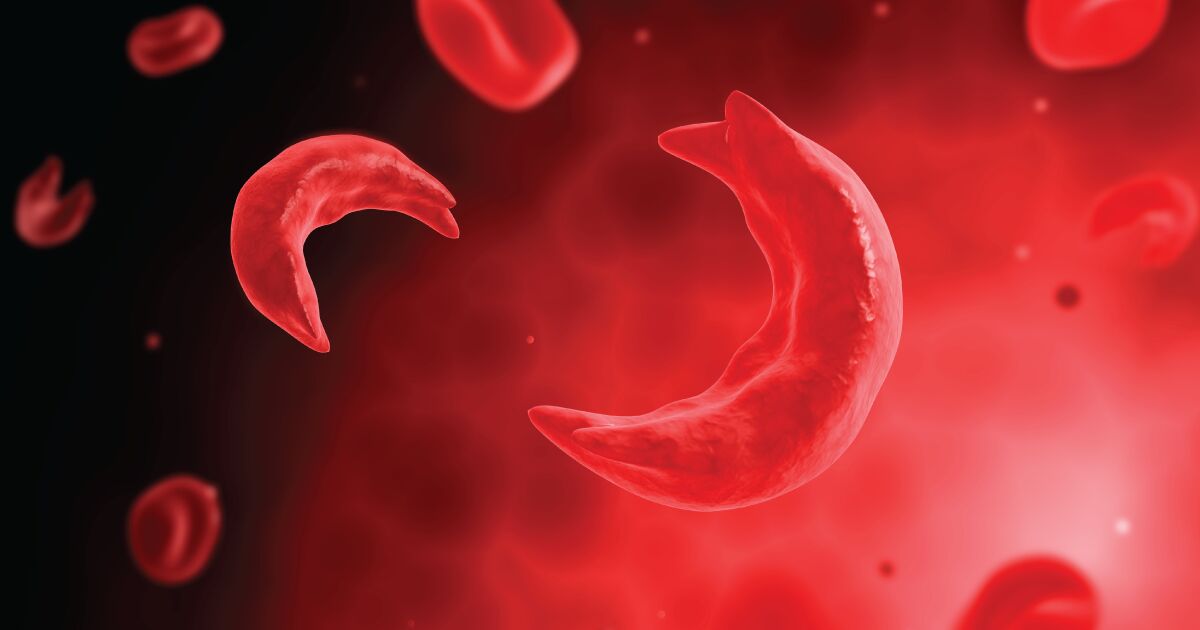
تهران - ایرنا - محققان راهکار جدیدی را برای درمان بیماری کم خونی داسیشکل و تالاسمی ابداع کردند که در آن دی ان ای سلولهای خون را با استفاده از فناوری ویرایش ژن CRISPR به طور دایم تغییر میدهند.
به گزارش روز دوشنبه از پایگاه خبری ساینس، در این تحقیقات حداقل 10 بیمار مبتلا به کم خونی داسی شکل و تالاسمی با استفاده از این شیوه جدید تحت درمان قرار گرفتند و چندین ماه است که بدون نیاز به روشهای درمانی متداول از قبیل انتقال خون مستمر و بدون تحمل درد به زندگی خود ادامه میدهند.
در دوران جنینی نوع خاصی از هموگلوبین تولید میشود که پس از تولد و شروع تنفس نوزاد، این هموگلوبین در اثر تغییر یک ژن با هموگلوبینهای بالغ جایگزین میشود. در افراد مبتلا به تالاسمی و کم خونی داسی شکل، این هموگلوبینهای بالغ دارای ایراد هستند.
اکنون محققان با استفاده از فناوری CRISPR توانستند ژن عامل تغییر هموگلوبین را بهگونهای ویرایش کنند که مجددا هموگلوبینهای جنین در بدن تولید شده و این بار به درستی به هموگلوبینهای بالغ تبدیل شوند.
هموگلوبین پروتئینی است که در گلبولهای قرمز خون وجود دارد و وظیفه آن انتقال اکسیژن و دی اکسید کربن است. هموگلوبین به طور طبیعی صاف و گرد است؛ گاهی هموگلوبین بسیار سخت، چسبنده و به شکل داس میشود و به راحتی از مویرگهای خونی عبور نمیکند. بنابراین اکسیژن به خوبی توسط خون حمل نمیشود و بیماری کمخونی داسی شکل بوجود میآید.
گاهی تغییر شکل و چسبندگی سلولها سبب میشود که رگهای خونی مسدود شوند و بیمار به دنبال درد شدید، در بیمارستان بستری شود. گاهی این عارضه به نارسایی اندام های داخلی بدن و مرگ زودهنگام منجر میشود. عفونت شدید، سکته مغزی، سردرد، مشکلات قلبی و کبدی از مهمترین عوارض کمخونی داسی شکل هستند.
بیماری سلول داسی شکل (SCD) یک بیماری ژنتیکی شایع است و آمار نشان میدهد که سالانه تقریبا 500 هزار نوزاد با این بیماری متولد میشوند و 50 درصد از این افراد قبل از پنج سالگی جان خود را از دست میدهند. این بیماری در همه نقاط دنیا شایع است؛ ولی در آفریقا و آسیا شیوع بیشتری دارد. در حال حاضر یکی از مهمترین روشهای درمان سلول داسی شکل، پیوند سلولهای بنیادی است.
تالاسمی یک اختلال خونی ژنتیکی است که سبب شکلگیری غیر طبیعی هموگلوبین و کم خونی میشود. این اختلال باعث تخریب و تضعیف سلولهای قرمز خون میشود. اختلال ژنتیکی تالاسمی از طریق ژنها از والدین به کودک به ارث میرسد. زمانی که یکی از والدین یا هر دوی آنان حامل این ژن باشند، احتمال ابتلای کودک نیز وجود دارد. در صورت ابتلای پدر و مادر به این بیماری، احتمال ابتلای فرزند بسیار بالاست.
انواع تالاسمی چهار نوعی اصلی تالاسمی وجود دارد که عبارتند از: تالاسمی آلفا تالاسمی بتا تالاسمی دلتا هموگلوبینوپاتی ترکیبی از این میان، انواع آلفا و بتا شایعتر هستند.
علل ابتلا به تالاسمی هنوز دلیل قطعی جهش ژنتیکی مرتبط با تالاسمی مشخص نیست.
شایع ترین علایم تالاسمی ضعف، خستگی، کندی رشد، تورم غیر طبیعی، صورت رنگ پریده، ساختار غیر طبیعی استخوان؛ به ویژه در ناحیه صورت و جمجمه، مشکلات قلبی و تجمع آهن در خون، مهمترین علایم این بیماری محسوب میشوند. علایم تالاسمی در برخی نوزادان کاملا مشهود است؛ در حالی که در برخی دیگر این علایم به مرور زمان در دو سال اول زندگی دیده میشود.
درمان بیماران مبتلا به تالاسمی شدید (ماژور)، نیازمند درمانهایی مانند پیوند مغز استخوان و دریافت خون هستند؛ در حالی که در بیماران مبتلا به تالاسمی خفیف (مینور)، ممکن است به درمان خاصی نیاز نباشد.
پیشگیری مهمترین روش برای جلوگیری از تولد نوزاد مبتلا به تالاسمی، آزمایش خون و آزمایشهای ژنتیکی قبل از ازدواج است که با دقت بالایی احتمال تولد نوزاد مبتلا را پیش بینی میکند. نتایج اولیه این تحقیقات در نشریه New England Journal of Medicine منتشر شده است.
*س_برچسبها_س*